3D Printed Plates for Cleft Lip and Palate Care in Less than Five Minutes
The treatment of children born with cleft lip and palate typically includes the use of therapeutic intraoral plates before surgical closure of the cleft. The plate passively sits on the roof of the baby’s mouth and helps separate the nasal and oral regions. So far, these plates have mostly been fabricated manually, and the process for their production still leaves room for improvement. Now, computer science researchers from ETH Zurich have teamed up with clinical care researchers at the University Hospital Basel to create a digital pipeline that enables the automatic production of the plates. The research project has promising potential to facilitate wider access to presurgical cleft treatment, especially in low-income countries.
About 1 in 700 children worldwide are born with cleft lip and palate. Orofacial clefts are the most common craniofacial malformations in newborns, and there are no existing effective preventive measures. While children in high-income countries with this birth condition receive appropriate care, children from low-income countries often lack this opportunity because treatment is expensive and requires high levels of medical and/or technical expertise. In addition, hospitals are often far away or not sufficiently equipped.
“These factors are especially relevant for the fabrication of presurgical orthopaedic plates, which are widely used for treating cleft lip and palate before surgery,” says Till Schnabel from ETH Zurich. Schnabel is a computer science doctoral researcher closely supervised by senior researchers Dr Baran Gözcü and Prof Barbara Solenthaler. They are part of ETH Zurich’s Computer Graphics Lab led by Prof Markus Gross. Schnabel and Gözcü are the first two authors of the research paper "Automated and data-driven plate computation for presurgical cleft lip and palate treatment" recently published in a special issue of the International Journal of Computer Assisted Radiology and Surgery. *
Within a few days after birth, a presurgical therapeutic plate designed to bridge the cleft space and to keep the baby from putting tongue pressure on the alveolar edges, i.e., the cleft opening is fitted to the baby’s mouth. The plate naturally facilitates the cleft opening becoming smaller after a few months, thus enabling a gentler repair surgery. The plate not only assists the infant in feeding but also fosters the baby’s development of early normal linguistic sounds.**
Conventionally, such a plate is fabricated using a plaster cast based on an imprint manually taken from the baby’s mouth. This intricate procedure requires highly trained specialists and carries some risk of the impression material entering the baby’s airways. More recently, handheld intraoral scanners have been introduced into practice to replace the imprint-taking, and – theoretically – to allow for fully digital workflows. However, most of these existing workflows and their 3D software are still too complex to be run in a non-specialised setting.
In contrast, the approach pursued by Schnabel and his colleagues at ETH Zurich, which was developed in collaboration with Andreas Mueller’s team at the University Hospital Basel, is as ambitious in development as it is straightforward in use: “Our aim has been to automate the whole process, which not only saves time but can also be done without an expert’s knowledge on site.”
At its core, the project employs a deep learning model, which was trained on a dataset that Schnabel and Mueller’s team assembled for this specific purpose. “Collecting sufficient data is often the greatest challenge when it comes to machine learning, especially for digital health, because medical data are sensitive and have to be treated confidentially and with the proper data security protocols,” says Till Schnabel. Nevertheless, they were able to gather around 400 scans: about two-thirds of children affected with a one-sided cleft and the other one-third of children with double clefts. “We collected data from old plaster cast imprints, which we digitized via 3D scanning, and we combined it with new data from intraoral scans,” explains Till Schnabel.
The deep learning model trained on this data can automatically recognise and select the principal structures, so-called landmarks, on the digital 3D model of the palate. In the rest of the pipeline, Schnabel and colleagues implemented established methods, such as surface registration and smoothing algorithms. “With the help of the selected landmarks, one can more accurately register a 3D scan, i.e., determine a close correspondence between the template and the input scan.” This method also provides a 3D segmentation differentiating various areas on the intraoral scan. The final step is automated plate computation, which comprises filling the cleft palate area and volumizing the surface to be 3D-printed. In short, these algorithms ensure that the plate is customised to the individual geometry of the baby’s mouth, and mimics, as best as possible, a healthy closed palatal shape.
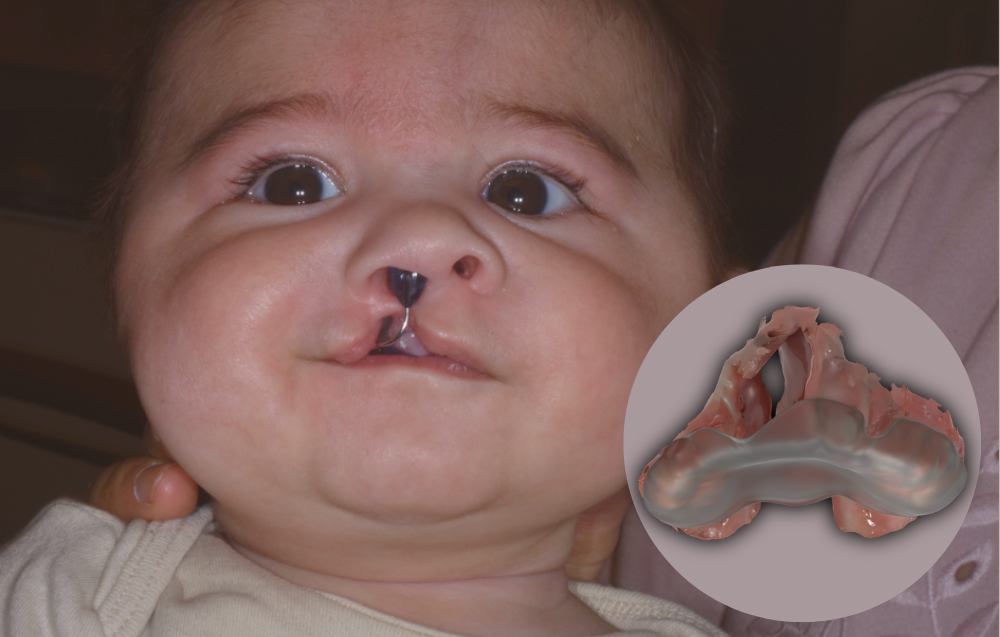
A baby wears the first palatal plate to be created entirely from the digital pipeline in Basel. As shown in the image, the plate is also fabricated with a small tab that nestles outside the mouth and helps to hold the plate in position. (Credit: Andreas Mueller and Benito Benitez). In the computer-generated rendering of the plate in the corner (Credit: Schnabel et al. 2023*), the palatal plate (translucent plastic) is shown overlaid on a baby’s intraoral scan of the unilateral cleft in the roof of the mouth. The plate effectively bridges the gap of the cleft and prevents the baby’s tongue from entering the intranasal region. View perspective is from the front of the baby’s mouth, gazing straight into the mouth towards the roof.
The pipeline has already been successfully implemented in the clinical routine in two hospitals, where 19 children have been treated with palatal plates that were produced with the digital pipeline. At the University Hospital Basel and Saveetha Medical College and Hospital, Chennai, India, digital intraoral scanning has already reduced the time necessary for conventional impression-taking from over 60 minutes to less than 20 minutes.
“The biggest difference, though, became evident in plate fabrication,” says Till Schnabel. “Previously, it would take close to an hour to make a plate – with our pipeline, the plate was done in less than 5 minutes.” Moreover, it didn’t require a plate specialist to operate complex software but only a few clicks on the computer. In other words, the plates are potentially accessible anywhere, regardless of facility resources. “In India, the workflow was made even more accessible. Hospitals did not need to have the printing infrastructure on site, the pipeline can be decentralised and is compatible with remote printing,” recounts Till Schnabel. “It worked perfectly.”
Schnabel is confident that the team’s automated pipeline will make a big difference in the treatment of cleft lip and palate, especially in low-income countries. “It has the potential to reduce the costs of cleft lip and palate care substantially,” he says, “making treatment accessible for those who might have been excluded so far.”
And thus, one has to emphasise the potential impact of the research. It can reduce the burden of this condition and the devastating effects it can have on children and their families; it can enable more children to heal.
Background
Till Schnabel is an early career researcher in a BRCCH research project “Burden-Reduced Cleft Lip and Palate Care and Healing", which is co-led by Prof Barbara Solenthaler (ETH Zurich) and Prof Andreas Mueller (University Hospital Basel and University of Basel). His work and the project are part of the overarching programme: Multi-Investigator Programme.
Interview article: Irène Dietschi
*Research paper
SCHNABEL TN, GÖZCÜ B, GOTARDO P, LINGENS L, DORDA D, Vetterli F, Emhemmed A, NALABOTHU P, LILL Y, BENITEZ BK, MUELLER AA, GROSS M & SOLENTHALER B. 2023. “Automated and Data-Driven Plate Computation for Presurgical Cleft Lip and Palate Treatment.” International Journal of Computer Assisted Radiology and Surgery. https://doi.org/10.1007/S11548-023-02858-6
**Reference
NALABOTHU P, BENITEZ BK, Dalstra M, Verna C, MUELLER AA. 2020. “Three-Dimensional Morphological Changes of the True Cleft under Passive Presurgical Orthopaedics in Unilateral Cleft Lip and Palate: A Retrospective Cohort Study”. Journal of Clinical Medicine. https://doi.org/10.3390/jcm9040962
Names in all caps indicate research contributions to the BRCCH consortium
Click here for a video abstract of the research paper